Tutorial¶
In this tutorial, we will identify the splicing derived neoantigens (both MHC bound T antigens and altered surface B antigens) on the TCGA Skin Cutaneous Melanoma (SKCM) cohort (472 tumor samples).
Note
We will use the whole dataset (472 bam files) to demostrate the full functionalities of SNAF. Completing this tutorial will take several hours and requires a multi-core High Performance Compute environment (HPC). Please replace the bam file folder with yours and configure your sample HLA type file as illustrated below.
Warning
There are a few steps in the workflow that requires internet connection, especially when launching B-antigen viewer. To avoid any errors, it is recommended to make sure your system has internet connection, which shouldn’t be a problem for any computer or HPC.
Running AltAnalyze to identify alternative splicing events¶
The analysis starts with a group of bam files, with each BAM corresponding to single patient sample. For example, if you have all the bam files stored in /user/ligk2e/bam,
the full path to the folder is the only command you need to run in this step (See Installation for more detail):
# run the container, the below command assume a folder named bam is in your current folder on the host machine
docker run -v $PWD:/usr/src/app/run -t frankligy123/altanalyze:0.5.0.1 bam
# if using singularity
singularity run -B $PWD:/usr/src/app/run --writable altanalyze/ bam
Warning
1. What if you only have one bam file? Our original Altanalyze codebase was designed for cohort-level splicing analysis (so at least 2 samples). As a workaround, a quick fix is to copy your bam file to another one (sample.bam, sample_copy.bam), put them all in a folder, then run the pipeline.
make sure there are no existing folders named
/bedand/altanalyze_outputin the same level of your/bamfolder.
The output of this step contains different useful readouts, including splicing junction quantification, splicing event quantification and gene expression, but the most important output that will be used
in the following step is the junction count matrix. The junction count matrix will be the altanalyze_output/ExpressionInput/counts.original.pruned.txt. Let us take a look at a subsampled junction count matrix, each row represent a splicing junction
annotated by the AltAnalyze gene model, and each column represents the sample name. The numerical value represents the number of reads that support the
occurence of a certain junction.
Note
In altanalyze/ExpressionInput folder, you will find counts.original.txt and counts.original.pruned.txt, in cohort level analysis (like dozens or hundreds)
of samples, we suggest using “pruned” one as additional filetering steps have been implemented to only keep splicing events that meet certain minimum read count (20)
and minimum frequency across cohort (>75%). In case where you only have < 10 samples, you may want to use “non-pruned” one to increase the sensitivity.
TCGA-EB-A82B-01A-11R-A352-07.bed |
TCGA-GF-A4EO-06A-12R-A24X-07.bed |
TCGA-D3-A1Q8-06A-11R-A18T-07.bed |
TCGA-FS-A1YY-06A-11R-A18T-07.bed |
TCGA-WE-A8ZM-06A-11R-A37K-07.bed |
TCGA-WE-A8ZN-06A-11R-A37K-07.bed |
TCGA-FW-A3TV-06A-11R-A239-07.bed |
TCGA-XV-AAZV-01A-11R-A40A-07.bed |
TCGA-DA-A1I1-06A-12R-A18U-07.bed |
TCGA-ER-A2NC-06A-11R-A18T-07.bed |
|
|---|---|---|---|---|---|---|---|---|---|---|
ENSG00000156398:E29.4-E31.1 |
1 |
1 |
0 |
2 |
3 |
0 |
3 |
0 |
2 |
3 |
ENSG00000095585:E8.1-E9.1 |
11 |
35 |
15 |
11 |
0 |
1 |
6 |
27 |
22 |
65 |
ENSG00000131069:E6.3-E10.2 |
39 |
99 |
98 |
71 |
64 |
9 |
86 |
49 |
77 |
34 |
ENSG00000171132:E14.1-E15.1 |
12 |
3 |
13 |
1 |
6 |
0 |
13 |
1 |
3 |
2 |
ENSG00000010278:E4.1-E6.1 |
69 |
5 |
146 |
149 |
54 |
27 |
47 |
68 |
13 |
26 |
ENSG00000214944:E40.1-E41.1 |
1 |
1 |
6 |
10 |
1 |
0 |
1 |
2 |
0 |
2 |
ENSG00000179912:E20.5-E27.1_57422959 |
0 |
2 |
4 |
2 |
0 |
0 |
2 |
0 |
2 |
3 |
ENSG00000186642:E9.2-E10.1 |
2 |
0 |
6 |
2 |
13 |
1 |
2 |
2 |
4 |
2 |
ENSG00000137497:E28.1-E29.1 |
265 |
110 |
95 |
221 |
349 |
157 |
200 |
111 |
221 |
115 |
ENSG00000114982:E22.1-E24.2 |
68 |
45 |
71 |
112 |
75 |
17 |
106 |
24 |
96 |
114 |
Note
In the future, we are planning to support user-supplied splicing count matrices from alternative algorithms, which will increase the compatability of the pipeline with other workflows.
Identify MHC-bound neoantigens (T-antigen)¶
With the junction count matrix, we can proceed to predict MHC-bound neoantigens (T antigen). The only additional input we need is
the patient HLA type information, in this analyis, we use Optitype to infer the 4 digit HLA type from RNA-Seq data, the sample_hla.txt file
looks like below example:
sample hla
TCGA-X1-A1WX-06A-11R-A38C-07.bed HLA-A*02:01,HLA-A*02:01,HLA-B*39:10,HLA-B*15:01,HLA-C*03:03,HLA-C*12:03
TCGA-X2-A1WX-06A-11R-A38C-07.bed HLA-A*02:01,HLA-A*01:01,HLA-B*40:01,HLA-B*52:01,HLA-C*03:04,HLA-C*12:02
TCGA-X3-A1WX-06A-11R-A38C-07.bed HLA-A*11:01,HLA-A*32:01,HLA-B*40:02,HLA-B*35:01,HLA-C*04:01,HLA-C*02:02
TCGA-X4-A2PB-01A-11R-A18S-07.bed HLA-A*02:01,HLA-A*01:01,HLA-B*07:02,HLA-B*18:01,HLA-C*07:01,HLA-C*07:02
Note
Optitype is again tool that is super easy to install as they provide the docker version, the input you need is the fastq file for your patient RNA-Seq sample, just follow their GitHub instructions. You can use your own favorite HLA typing tool as well. All we need is the HLA typing information. I provide the Optitype script I used here: 3. How to do HLA genotyping?.
Loading and instantiating¶
Load the packages:
import os,sys
import pandas as pd
import numpy as np
import snaf
The first step is to load our downloaded reference data into the memory to facilitate the repeated retrieval of the data while running:
# database directory (where you extract the reference tarball file)
db_dir = '/user/ligk2e/download'
# instantiate (if using netMHCpan)
netMHCpan_path = '/user/ligk2e/netMHCpan-4.1/netMHCpan'
snaf.initialize(db_dir=db_dir,gtex_mode='count',binding_method='netMHCpan',software_path=netMHCpan_path)
# instantiate (if not using netMHCpan)
snaf.initialize(db_dir=db_dir,gtex_mode='count',binding_method='MHCflurry',software_path=None)
Note
Explaination of gtex_mode argument: We provide two ways for GTEx filtering, one is using splicing junction count (gtex_mode='count'),
The another is using splicing percent spliced in (PSI) so that gtex_mode='psi', since in this tutorial we are using splicing junction
count matrix as the quantification of the splicing junction, we call the count mode for GTEx filtering. We allow user to supply a PSI
matrix, in this case, you should set gtex_mode='psi'.
Running the T antigen workflow¶
We first instantiate JunctionCountMatrixQuery object, here the df is the junction count matrix (a pandas dataframe) that we refer to above.:
df = pd.read_csv('altanalyze_output/ExpressionInput/counts.original.pruned.txt',sep='\t',index_col=0)
jcmq = snaf.JunctionCountMatrixQuery(junction_count_matrix=df)
We will parse the HLA type sample_hla.txt file into a nested list. The goal is to have a python nested list hlas, where each element in
hlas is another list, for example [HLA-A*02:01,HLA-A*02:01,HLA-B*39:10,HLA-B*15:01,HLA-C*03:03,HLA-C*12:03]. Make sure the order of the element is consistent
with the sample order present in the column of junction count matrix. In another words, if the column of junction matrix is “sample1,sample2,sample3,..”,
then make sure the first element in hlas is the HLA type for sample1, then sample2, sample3:
sample_to_hla = pd.read_csv('sample_hla.txt',sep='\t',index_col=0)['hla'].to_dict()
hlas = [hla_string.split(',') for hla_string in df.columns.map(sample_to_hla)]
Note
The above step depends on how your HLA typing file looks like, so just adjust it accordingly.
The main program can be wrapped into one line of code. A folder named result will be created and the resultant JunctionCountMatrixQuery
object will be saved as a pickle file:
jcmq.run(hlas=hlas,outdir='./result')
To generate a series of useful outputs including neoantigen burden and neoantigen frequency, we deserialize the pickle file back to memory and automatically generate these output files:
snaf.JunctionCountMatrixQuery.generate_results(path='./result/after_prediction.p',outdir='./result')
Now in the result folder, we can have neoantigen burden files associated with each stage of the workflow. A stage
refers to different stages in the neoantigen production process, first and foremost, a neoantigen is derived from a neojunction (splicing event), then all potential
peptides will be generated after in-silico translation, followed by MHC presentation and MHC-peptide complex formation to elicit a T cell response. We argue that exporting
neoantigens at each stages are useful for various downstream analyses.
stage 0: neojunction, the number of tumor-specific junction readsstage 1: peptides that are predicted (3-way in-silico translation) from each neojunctionstage 2: peptides that are predicted to be presented on an MHC molecule (based on netMHCpan or MHCflurry prediction)stage 3: peptides that are predicted to be immunogenic (DeepImmuno)
For each stage, you may see the following categories of results:
burden_stage{0-3}.txt: This file characterizes the patient level neoantigen burden (See below concrete example).frequency_stage{0-3}.txt: This file chracterizes each specific neoantigen, how many times does it occur across the whole cohort?frequency_stage{0-3}_verbosity1_uid.txt: This is an enhanced version of frequency.txt file, where each row contains both the neoantigen and the source junction uid. This file can be further enhanced by adding Add gene symbol and Add chromsome coordinate. See Compatibility (Gene Symbol & chromsome coordinates).x_neoantigen_frequency{0-3}.pdf: This is a visual representation of neoantigen frequency as a sorted barplot, where each bar is a neoantigen and the height is its occurence across cohorts.x_occurence_frequency{0-3}.pdf: This is an alternative visualization of neoantigen frequency as a histplot, interval (x-axis) with the occurence of each neoantigen across the cohort.
The burden matrix should look like the below, where the last column and last row represent the mean burden for each feature and the total burden for each sample. Since this output only illustrates the last 10 columns and rows, all of the entries are zero, to give the user a sense of the file layout.
TCGA-DA-A3F3-06A-11R-A20F-07.bed |
TCGA-EE-A3JI-06A-11R-A21D-07.bed |
TCGA-EE-A2GU-06A-11R-A18T-07.bed |
TCGA-EE-A2GB-06A-11R-A18T-07.bed |
TCGA-EE-A29N-06A-12R-A18S-07.bed |
TCGA-FS-A1ZK-06A-11R-A18T-07.bed |
TCGA-EE-A3AG-06A-31R-A18S-07.bed |
TCGA-EE-A2GD-06A-11R-A18T-07.bed |
TCGA-EE-A2MD-06A-11R-A18T-07.bed |
mean |
|
|---|---|---|---|---|---|---|---|---|---|---|
ENSG00000112208:E1.1-E2.1 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E6.1-E10.1 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E20.1-E21.2 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E2.11-E3.3 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E2.11-E3.2 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E19.2-E20.1 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E18.1-E19.2 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000112200:E17.1-E18.1 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
ENSG00000125676:E31.2-E32.1 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
0.0 |
burden |
5601.0 |
5641.0 |
5653.0 |
5693.0 |
5716.0 |
5949.0 |
6222.0 |
6418.0 |
6700.0 |
2858.5 |
Neoantigen frequency plot shows the distinctive pattern between shared neoantigens (left part) and unique neoantigens (right part).
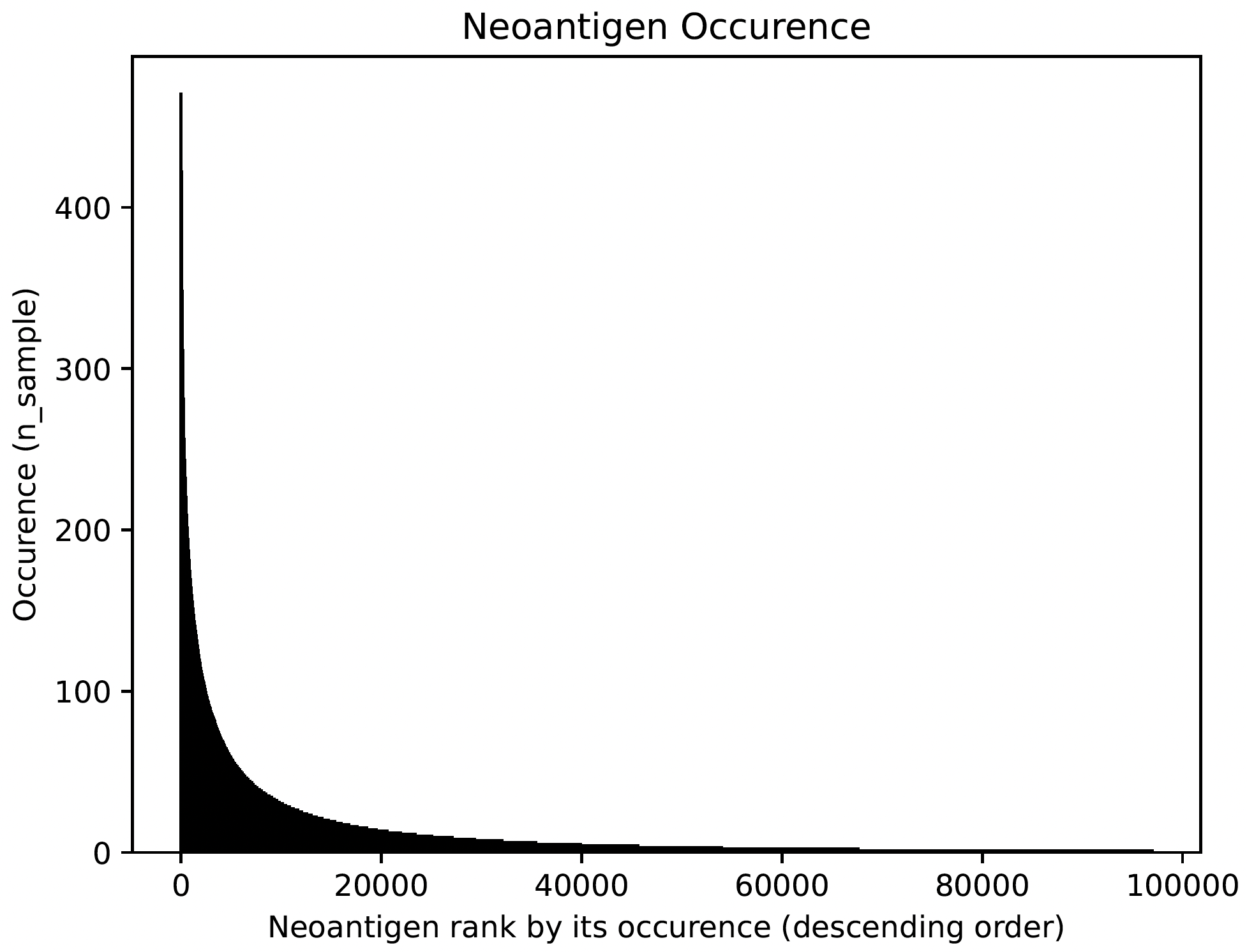
Users can also report T cell Neoantigen associated with a speficic sample (precision medicine) by running report_candidates. The candidates reported will look like the below table:
sample |
peptide |
uid |
hla |
binding_affinity |
immunogenicity |
n_sample |
|---|---|---|---|---|---|---|
SRR5933726.Aligned.sortedByCoord.out.bed |
SLAPQPLAL |
ENSG00000181143:E138.1-E139.1 |
HLA-C*02:02 |
0.267 |
0.9917507171630859 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
SLAPQPLAL |
ENSG00000181143:E138.1-E139.1 |
HLA-C*16:01 |
0.403 |
0.9909228086471558 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
RSESHPRAL |
ENSG00000124193:E2.1-I2.1 |
HLA-C*02:02 |
0.754 |
0.6034059524536133 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
RSESHPRAL |
ENSG00000124193:E2.1-I2.1 |
HLA-C*16:01 |
0.098 |
0.43916165828704834 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
NGFTHQSSM |
ENSG00000181143:E141.1-E142.1 |
HLA-C*02:02 |
1.561 |
0.7201588153839111 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
NGFTHQSSM |
ENSG00000181143:E141.1-E142.1 |
HLA-C*16:01 |
0.521 |
0.7946109771728516 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
NGFTHQSSM |
ENSG00000181143:E141.1-E142.1 |
HLA-B*51:01 |
1.468 |
0.9217257499694824 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
TASPLLVLF |
ENSG00000181143:E143.1-E144.1 |
HLA-C*02:02 |
0.012 |
0.9696042537689209 |
14 |
SRR5933726.Aligned.sortedByCoord.out.bed |
TASPLLVLF |
ENSG00000181143:E143.1-E144.1 |
HLA-C*16:01 |
0.081 |
0.9623821973800659 |
14 |
Interface to proteomics validation¶
Now imagine we have a handful of predicted short-peptides that potentially can be therapeutically valuable targets, as a routine step, we definitely want to test whether they are supported by public or in-house MS (either untargeted or targetted HLA-bound immunopeptidome) datasets. We provide a set of functions that can make this validation process easier.
First, we want to extract all candidate and write them into a fasta file:
jcmq = snaf.JunctionCountMatrixQuery.deserialize('result/after_prediction.p')
sample = 'SRR5933735.Aligned.sortedByCoord.out'
jcmq.show_neoantigen_as_fasta(outdir='./fasta',name='neoantigen_{}.fasta'.format(sample),stage=2,verbosity=1,contain_uid=True,sample=sample)
Then, we want to remove identical peptides, becasue same peptide can be generated from different junctions:
snaf.proteomics.remove_redundant('./fasta/neoantigen_{}.fasta'.format(sample),'./fasta/neoantigen_{}_unique.fasta'.format(sample))
Next, we want to remove all peptides that are overlapping with human proteome, you can download any preferred human proteome database (UCSC or Uniprot), we provide a reference fasta human_uniprot_proteome.fasta downloaded from Uniprot downloaded at Jan 2020, available at Synapse, we chop them into 9 and 10 mers without duplicates. Then we remove overlapping candidates:
chop_normal_pep_db(fasta_path='human_uniprot_proteome.fasta',output_path='./fasta',mers=[9,10],allow_duplicates=False)
# human_proteome_uniprot_9_10_mers_unique.fasta is generated from command above
snaf.proteomics.compare_two_fasta(fa1_path='./fasta/human_proteome_uniprot_9_10_mers_unique.fasta',
fa2_path='./fasta/neoantigen_{}_unique.fasta'.format(sample),outdir='./fasta',
write_unique2=True,prefix='{}_'.format(sample))
# here write_unique2 means we only report peptides that are unique to second fasta file, which is our candidates fasta files.
Usually, MS software requires a customized fasta database, you’ve already had that right now. Depending on which MS software you use, the configuration steps can vary, but we recommend using MaxQuant here which is highly regarded. MaxQuant requires to compile a configuration files called mqpar.xml which stores the setting for the search engine, the databases that will be used and the input raw files, manually adjusting it can be a pain, so we provide a handy function to automatically do so:
dbs = ['/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/RNA/snaf_analysis/fasta/SRR5933726.Aligned.sortedByCoord.out.bed_unique2.fasta']
inputs = ['/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48/OvCa48_classI_Rep#1.raw',
'/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48/OvCa48_classI_Rep#2.raw',
'/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48/OvCa48_classI_Rep#3.raw',
'/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48/OvCa48_classI_Rep#4.raw',
'/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48/OvCa48_classI_Rep#5.raw',
'/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48/OvCa48_classI_Rep#6.raw']
outdir = '/data/salomonis2/LabFiles/Frank-Li/neoantigen/MS/schuster/MS/OvCa48'
snaf.proteomics.set_maxquant_configuration(dbs=dbs,n_threads=20,inputs=inputs,enzymes=None,enzyme_mode=5,protein_fdr=1,peptide_fdr=0.05,site_fdr=1,
outdir=outdir,minPepLen=8,minPeptideLengthForUnspecificSearch=8,maxPeptideLengthForUnspecificSearch=25)
A automatically generated configuration file (mqpar.xml) will be shown in the outdir that you specified. More information can be found in the Interface to proteomics.
Visualization¶
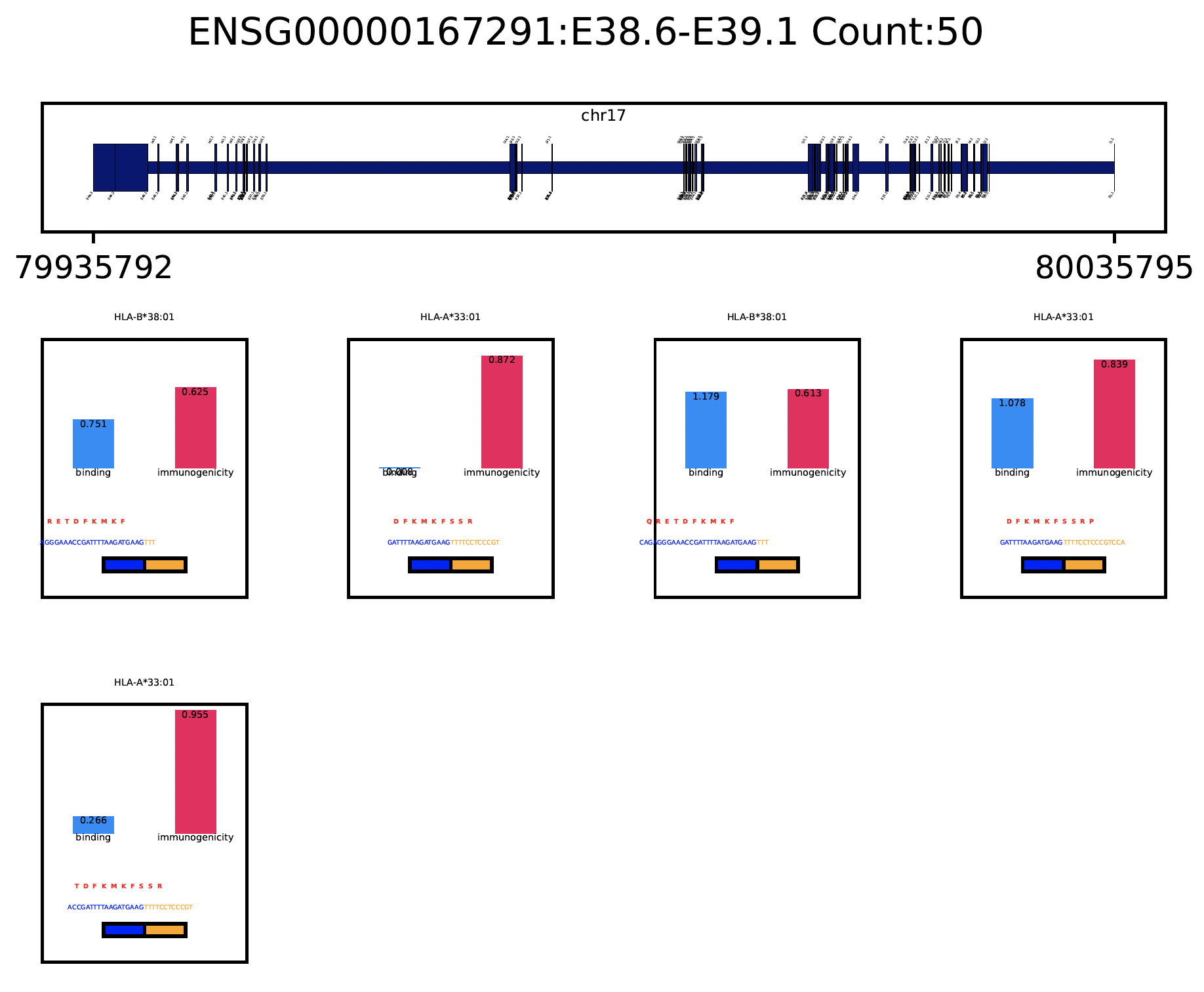
A very important question users will want to ask is what splicing event produces a certain neoepitope? We provide a convenient plotting function to achieve this, usually we want to first deserialize the resultant pickle object back to memory from last step:
jcmq = snaf.JunctionCountMatrixQuery.deserialize('result/after_prediction.p')
jcmq.visualize(uid='ENSG00000167291:E38.6-E39.1',sample='TCGA-DA-A1I1-06A-12R-A18U-07.bed',outdir='./result')
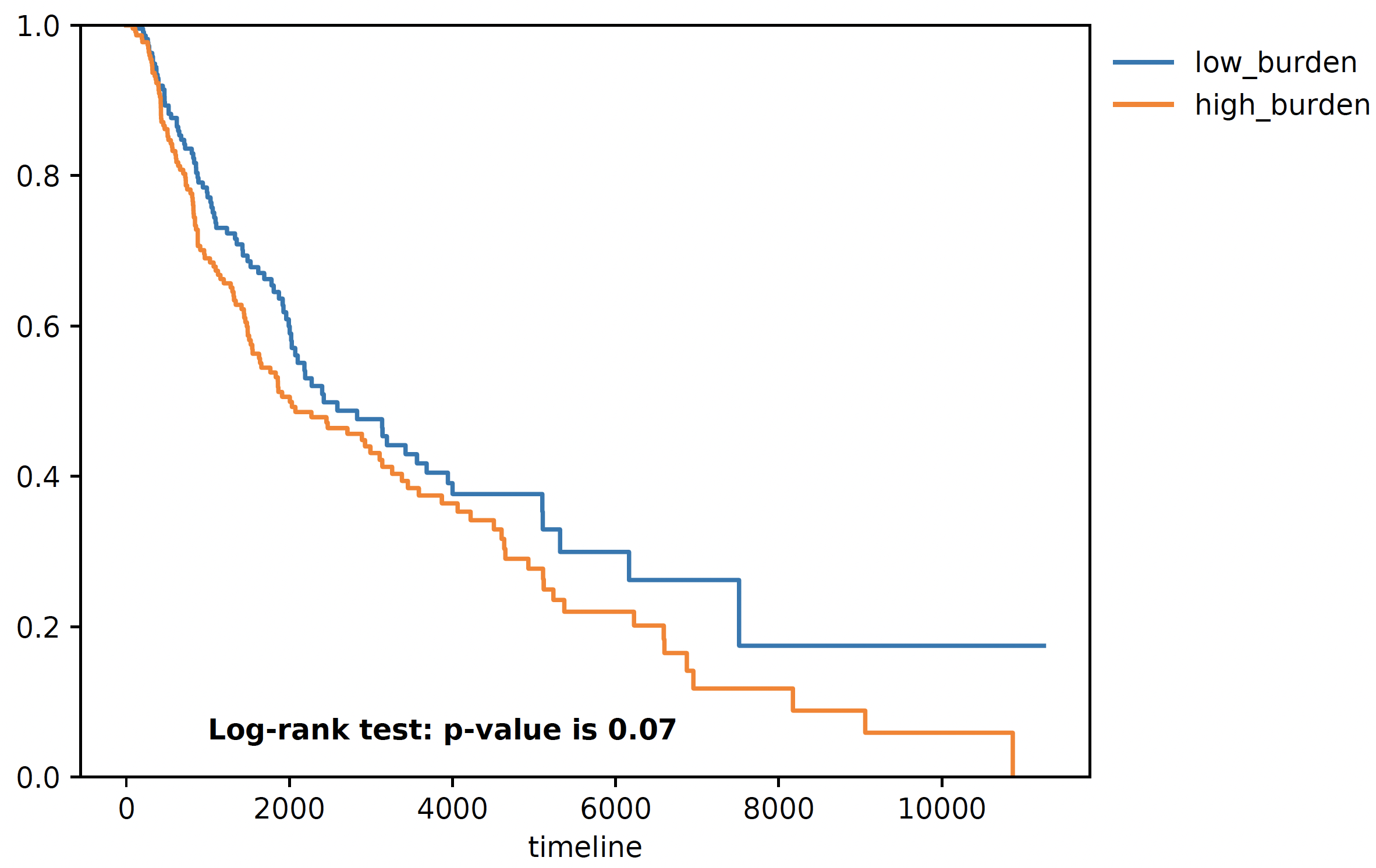
Survival Analysis¶
We download the TCGA SKCM survival data from Xena browser, we provide a convenient function to do a survival analyis using various stratification criteria, To use this function, we need a dataframe (survival) whose index is sample name, along with two columns one representing event (OS.death) and one representing duration (OS.time). Another is burden, it is a pandas series with sample name as index, and neoantigen burden as values. The sample name needs to be the same, that’s why we need a few lines of code for parsing below:
survival = pd.read_csv('TCGA-SKCM.survival.tsv',sep='\t',index_col=0) # 463
burden = pd.read_csv('result/burden_stage2.txt',sep='\t',index_col=0).loc['burden',:].iloc[:-1] # 472
burden.index = ['-'.join(sample.split('-')[0:4]) for sample in burden.index]
# convenient function for survival
snaf.survival_analysis(burden,survival,n=2,stratification_plot='result/stage2_stratify.pdf',survival_plot='result/stage2_survival.pdf')
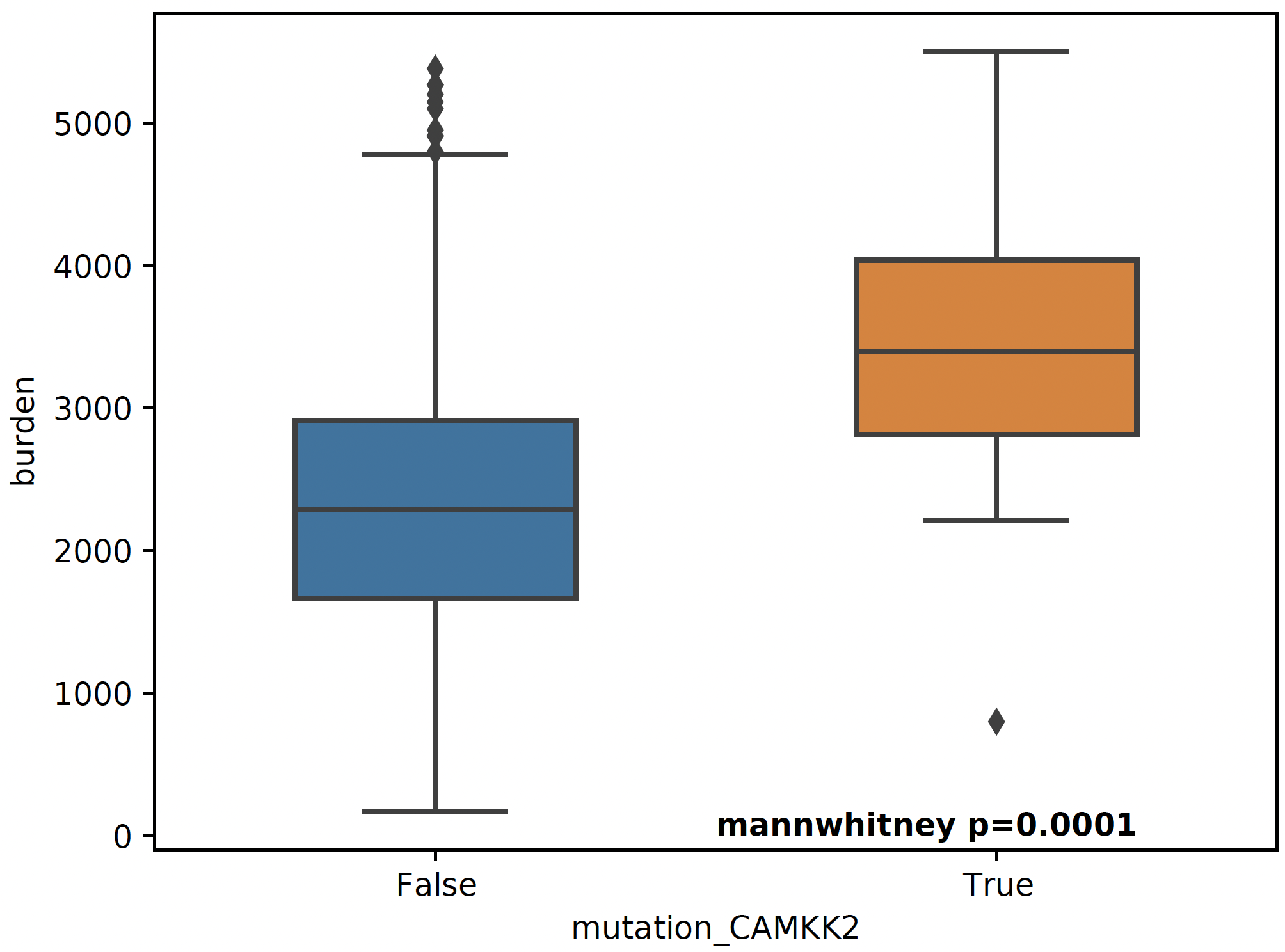
Mutation Association Analysis¶
We download the TCGA SKCM mutation data from <Xena browser>. We provide a convenient function to calculate all associations and plot them. To explain how
this function work, basically, it has two mode, compute mode is to compute the association between each gene mutation and neoantigen burden. plot mode
is to visualize selective genes as a side-by-side barplot. For compute mode, we need the burden file (again, a pandas series, same as described above in survival analysis),
and mutation, which is a dataframe whose index is sample name, and one column represents mutated gene. For plot mode, just need to specify a list of
genes to plot:
mutation = pd.read_csv('TCGA-SKCM.mutect2_snv.tsv',sep='\t',index_col=0) # 467 samples have mutations
mutation = mutation.loc[mutation['filter']=='PASS',:]
burden = pd.read_csv('result/burden_stage3.txt',sep='\t',index_col=0).loc['burden',:].iloc[:-1] # 472
burden.index = ['-'.join(sample.split('-')[0:4]) for sample in burden.index]
# mutation convenience function, compute mode
snaf.mutation_analysis(mode='compute',burden=burden,mutation=mutation,output='result/stage3_mutation.txt',gene_column='gene')
# mutation convenience function, plot mode
snaf.mutation_analysis(mode='plot',burden=burden,mutation=mutation,output='result/stage3_mutation_CAMKK2.pdf',genes_to_plot=['CAMKK2'])
mutation_gene |
n_samples |
pval |
adjp |
|---|---|---|---|
OGFOD3 |
13 |
3.82E-05 |
0.272783716 |
CAMKK2 |
19 |
5.25E-05 |
0.272783716 |
ACE |
56 |
0.00305804 |
0.357487136 |
PALMD |
22 |
0.003794017 |
0.357487136 |
CEP350 |
35 |
0.001240149 |
0.357487136 |
PROM2 |
50 |
0.000457734 |
0.357487136 |
STAT4 |
40 |
0.003968346 |
0.357487136 |
DOCK10 |
24 |
0.000289835 |
0.357487136 |
QARS |
12 |
0.000546601 |
0.357487136 |
For a specific mutation CAMKK2, which has been reported that the suppresses expression of this gene, which can increase ferroptosis efficacy and
anti-PD1 immunotherapy (paper link), we show that patients with mutated CAMKK2 have higher
neoantigen burden so that can explain why it lead to better immunotherapy efficacy.
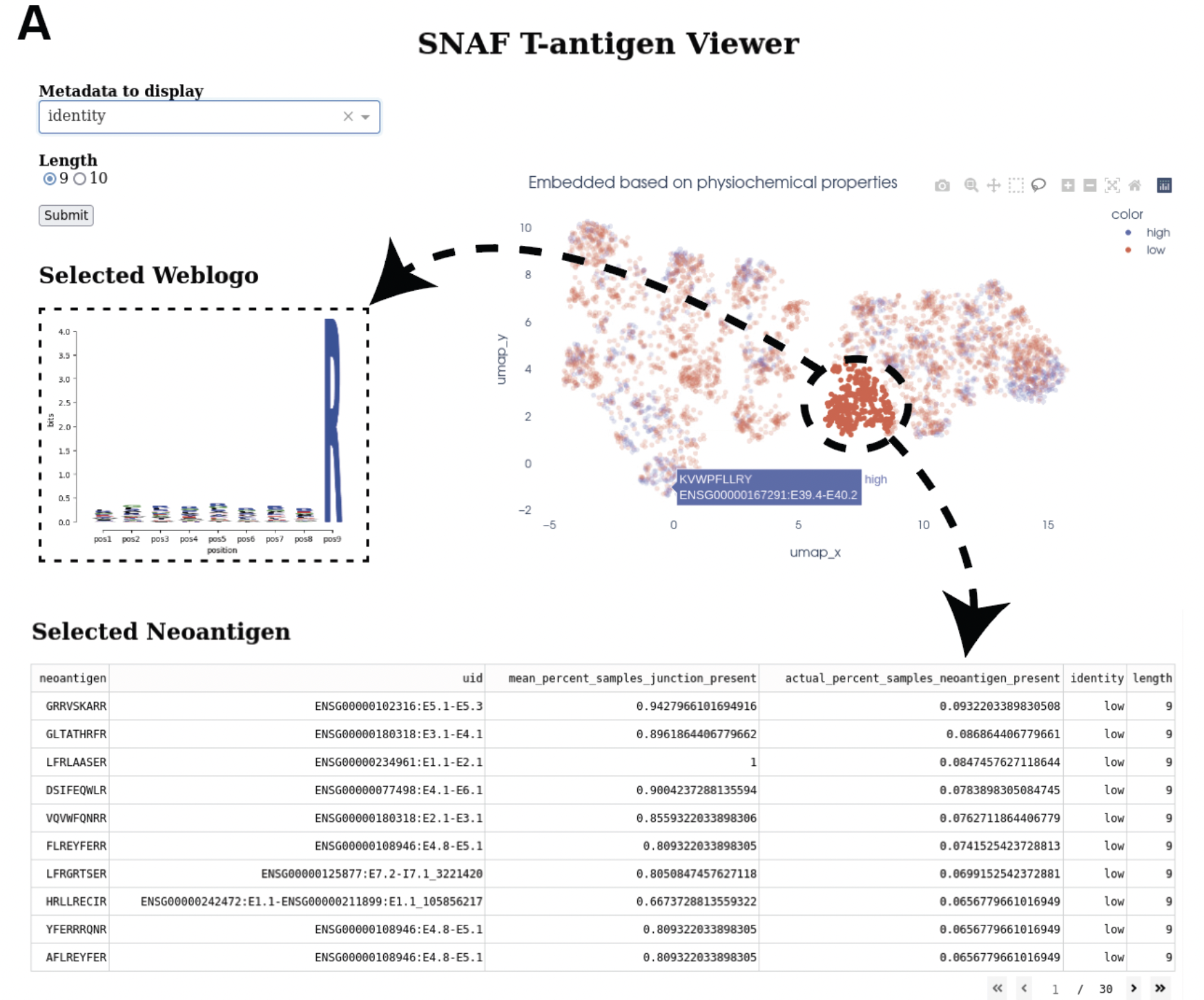
Interactive Neoantigen Viewer¶
Users can launch a dash interactive neoantigen viewer to visualize all the neoantigens based on their physiochemical properties and their motif
composition along with the source splicing junction. To achieve it, we first run a pre-processing step analyze_neoantigens to generate
some portable input file for the viewer, we need a file named shared_vs_unique_neoantigen_all.txt. Be sure the specify the full name for this file,
also, the umap plot may take 10 seconds to load if you don’t see it loads instantly:
snaf.analyze_neoantigens(freq_path='result/frequency_stage2_verbosity1_uid.txt',junction_path='result/burden_stage0.txt',total_samples=2,outdir='result',mers=None,fasta=False)
snaf.run_dash_T_antigen(input_abs_path='/data/salomonis2/LabFiles/Frank-Li/neoantigen/TCGA/SKCM/snaf_analysis/result/shared_vs_unique_neoantigen_all.txt')
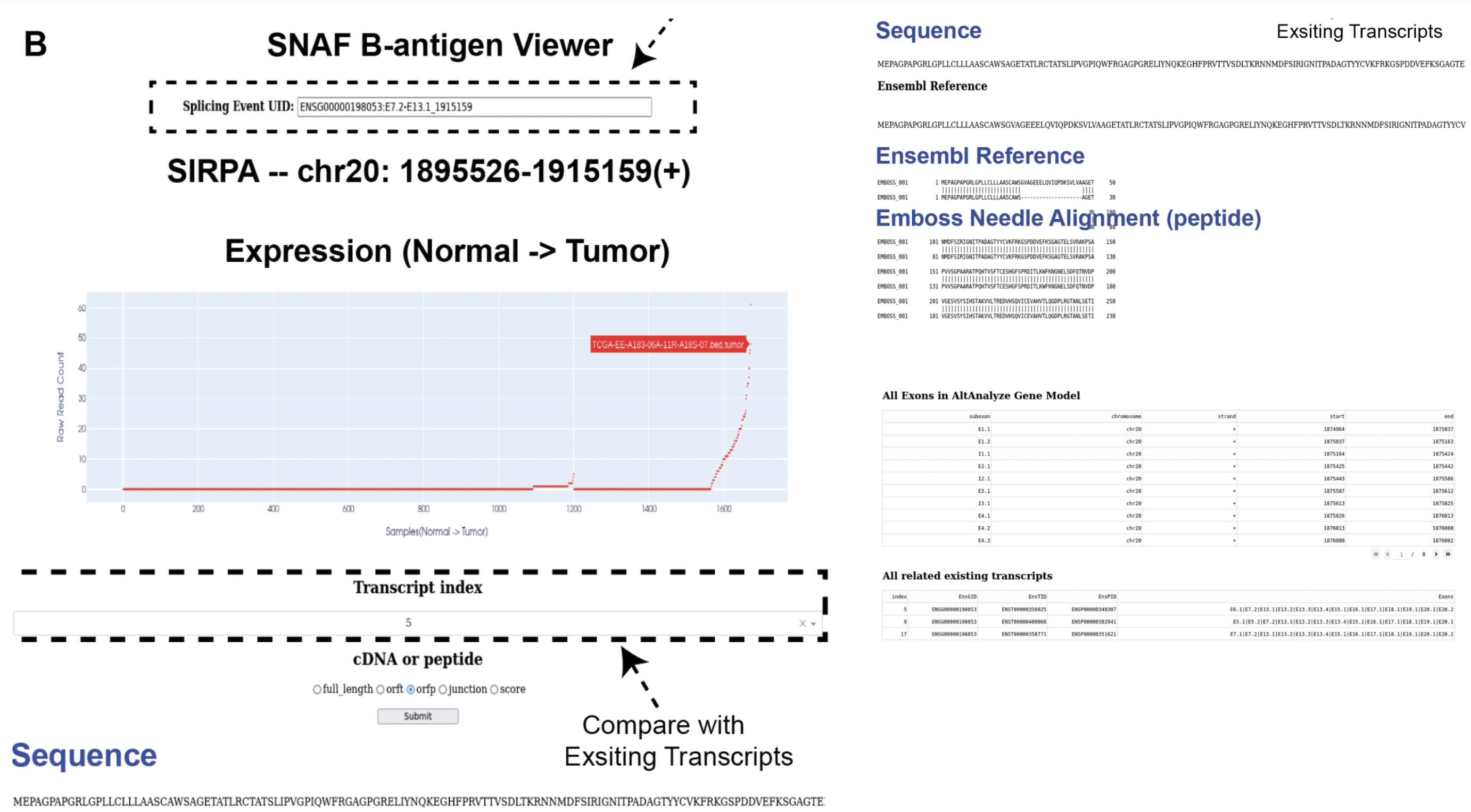
Identify altered surface proteins (B-antigen)¶
As a separate workflow, the B-antigen pipeline aims to priotize the altered surface protein from abnormal splicing events.
Instantiating B pipeline¶
We again load some necessary reference data files to RAM:
# same as T antigen pipeline
import snaf
import pandas
db_dir = '/user/ligk2e/download'
netMHCpan_path = '/user/ligk2e/netMHCpan-4.1/netMHCpan'
snaf.initialize(db_dir=db_dir,gtex_mode='count',binding_method='netMHCpan',software_path=netMHCpan_path)
# additional instantiation steps
from snaf import surface
surface.initialize(db_dir=db_dir)
Running the program¶
We first obtain the membrane splicing events:
df = pd.read_csv('altanalyze_output/ExpressionInput/counts.original.pruned.txt',sep='\t',index_col=0)
membrane_tuples = snaf.JunctionCountMatrixQuery.get_membrane_tuples(df)
Then we run the B pipeline:
# if using TMHMM
surface.run(membrane_tuples,outdir='result',tmhmm=True,software_path='/data/salomonis2/LabFiles/Frank-Li/python3/TMHMM/tmhmm-2.0c/bin/tmhmm')
# if not using TMHMM
surface.run(membrane_tuples,outdir='result',tmhmm=False,software_path=None)
After this step, a pickle file will again be deposited to the result folder. However, we do want to generate human-readable results:
# if having gtf file for long-read data
surface.generate_results(pickle_path='./result/surface_antigen.p',outdir='result',strigency=5,gtf='./SQANTI-all/collapse_isoforms_classification.filtered_lite.gtf')
# if not having
surface.generate_results(pickle_path='./result/surface_antigen.p',outdir='result',strigency=3,gtf=None)
Different strigencies are explanined below:
strigency 1: The novel isoform needs to be absent in UniProt databasestrigency 2: The novel isoform also needs to be a documented protein-coding genestrigency 3: The novel isoform also needs to not be subjected to Nonsense Mediated Decay (NMD)strigency 4: The novel isoform also needs to have long-read or EST support (as long as the novel junction present in full-length)strigency 5: The novel isoform also needs to have long-read or EST support (whole ORF needs to be the same as full-length)
An output called candidates.txt is what we are looking for, to facilitate the inspection of the result, let’s use the B antigen viewer shown below. Also,
we can generate a more readable and publication-quality table for the candidate by using report_candidates.
Interactive neoantigen viewer¶
Similar to T antigen, users can explore all the altered surface protein for B antigen, we need the pickle object and the candidates file,
importantly, please specify the full path to the python executable you use to run your python script:
surface.run_dash_B_antigen(pkl='result/surface_antigen.p',candidates='result/candidates_5.txt',
python_executable='/data/salomonis2/LabFiles/Frank-Li/refactor/neo_env/bin/python3.7')
Note
The reason for specifying python_executable is for using EmBoss Needleman global alignment REST API. As the REST API was provided as a python script, I need the python executable full path to execute the script.
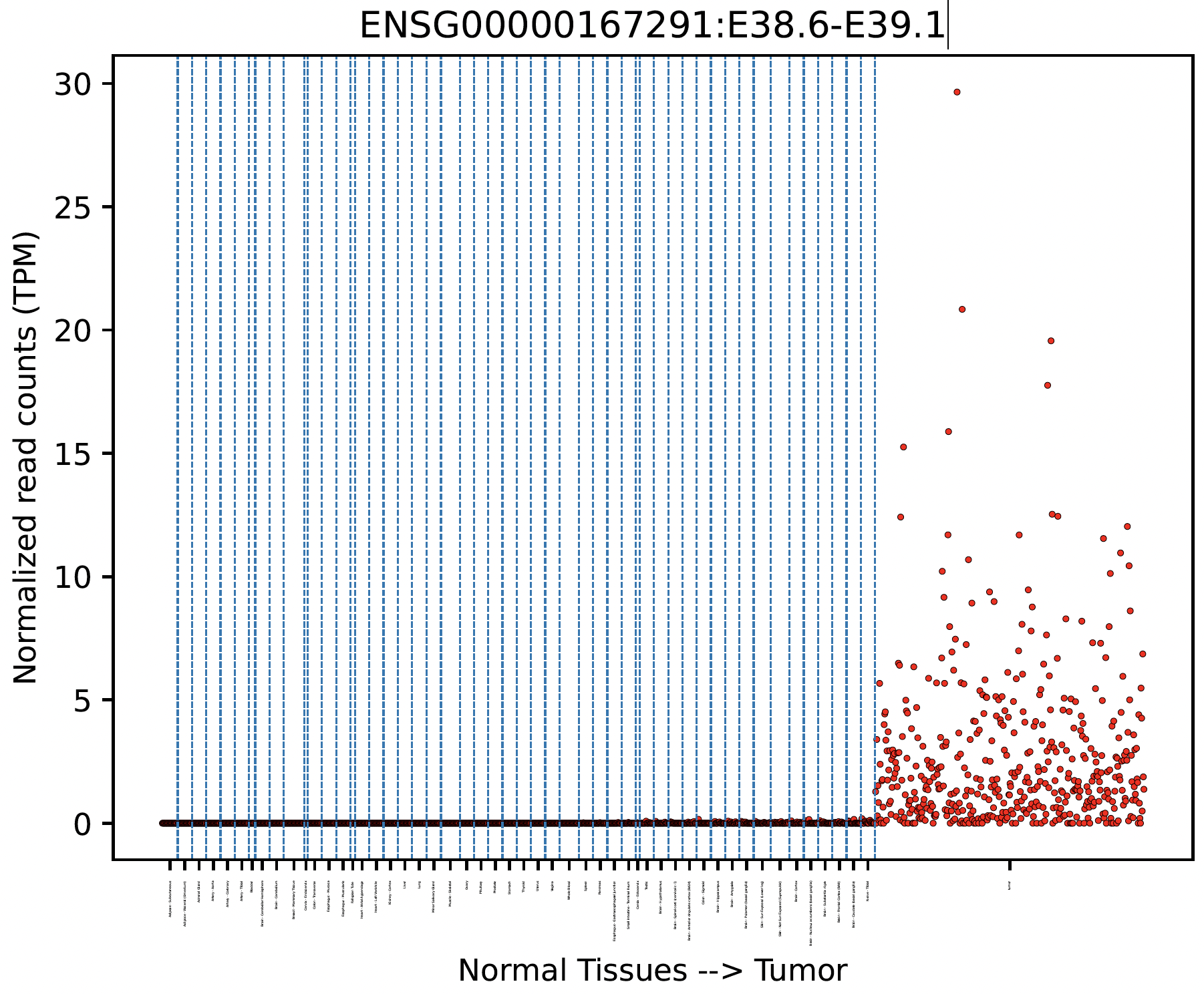
Tumor Specificity (GTEx)¶
For a specific splicing event, we can visualize its tumor specificity by comparing its expression in tumor versus normal tissue:
snaf.gtex_visual_combine('ENSG00000167291:E38.6-E39.1',norm=True,outdir='result',tumor=df)
here norm argument controls whether to normalize the raw read count to Count Per Million (CPM) to account for sequencing depth bias.
You can also view each tissue type separately:
snaf.gtex_visual_subplots('ENSG00000198053:E7.2-E13.1_1915159',norm=True,outdir='result')
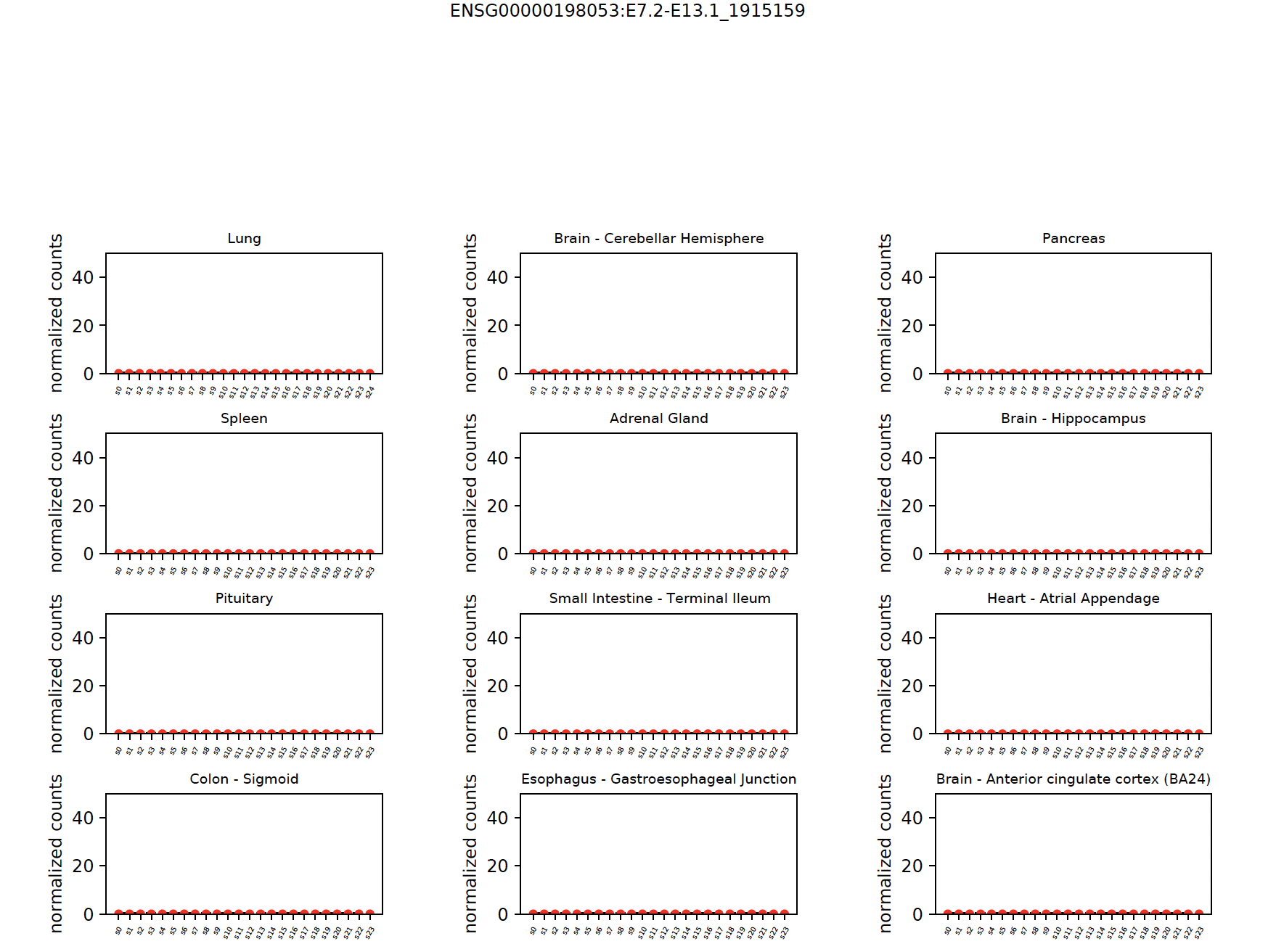
Note
SNAF also provide quantitative measurement for tumor specificity, to calculate the tumor specificity for each neojunction, we need to run Add tumor specificity scores. We can report mean GTEx read count, Maximum likelihood Estimation and hierarchical Bayesian estimation, the detailed mathematical equations are shown in the preprint.
Compatibility (Gene Symbol & chromsome coordinates)¶
For some historical reasons, different RNA splicing pipeline (i.e. AltAnalyze, MAJIQ, rMATs, LeafCutter, etc) use their own gene model, meaning how they define and index gene and exon number. Hence, a splicing junction (chromsome coordinate like chr7:156999-176000) maybe reprensented in diverse annotation in different pipelines.
It is in our to-do list but also requires a lot of work to harmonize all the annotations, for now, we provide functions to convert AltAnalyze annotation
to the most generic representation, namely, gene symbol and chromosome coordinates. It will be handled by two functions, Add gene symbol and Add chromsome coordinate.
Now let’s take the output frequency_stage2_verbosity1_uid.txt as the example (most important thing is pandas dataframe index format):
n_sample
TQLSVPWRL,ENSG00000258017:E2.3-E2.6 470
QIFESVSHF,ENSG00000198034:E8.4-E9.1 463
MGSKRLTSL,ENSG00000241343:E2.2-E2.4 449
HALLVYPTL,ENSG00000090581:E5.10-E5.24 435
QFADGRQSW,ENSG00000111843:E9.1-ENSG00000137210:E6.1 433
GIHPSKVVY,ENSG00000263809:E3.1-E4.1 432
RPYLPVKVL,ENSG00000134330:E8.4-E9.1 432
LPPPRLASV,ENSG00000090581:E5.10-E5.24 428
SSQVHLSHL,ENSG00000172053:E11.8-E11.11 425
Let’s add gene symbol to the dataframe:
df = snaf.add_gene_symbol_frequency_table(df=df,remove_quote=True)
Note
The remove_quote argument is due to the fact that in frequency.txt file, one column is the list of all sample names that contain
the splicing neoantigen. The thing is, when such a list being re-read into the memory, sometimes a quotation will be added so that the data type
become a string instead of list, which is not desirable, so if your df is read using pd.read_csv, you need to set it as True,
otherwise, set it as False.
The resultant will look like that:
n_sample symbol
TQLSVPWRL,ENSG00000258017:E2.3-E2.6 470 unknown_gene
QIFESVSHF,ENSG00000198034:E8.4-E9.1 463 RPS4X
MGSKRLTSL,ENSG00000241343:E2.2-E2.4 449 RPL36A
HALLVYPTL,ENSG00000090581:E5.10-E5.24 435 GNPTG
QFADGRQSW,ENSG00000111843:E9.1-ENSG00000137210:E6.1 433 TMEM14C
GIHPSKVVY,ENSG00000263809:E3.1-E4.1 432 unknown_gene
RPYLPVKVL,ENSG00000134330:E8.4-E9.1 432 IAH1
LPPPRLASV,ENSG00000090581:E5.10-E5.24 428 GNPTG
SSQVHLSHL,ENSG00000172053:E11.8-E11.11 425 QARS1
Let’s add chromsome coorinates to the splicing junction annotation as well:
df = snaf.add_coord_frequency_table(df=df,remove_quote=False)
Results look like this:
n_sample symbol coord
TQLSVPWRL,ENSG00000258017:E2.3-E2.6 470 unknown_gene chr12:49128207-49128627(+)
QIFESVSHF,ENSG00000198034:E8.4-E9.1 463 RPS4X chrX:72256054-72272640(-)
MGSKRLTSL,ENSG00000241343:E2.2-E2.4 449 RPL36A chrX:101391235-101391459(+)
HALLVYPTL,ENSG00000090581:E5.10-E5.24 435 GNPTG chr16:1362320-1362452(+)
QFADGRQSW,ENSG00000111843:E9.1-ENSG00000137210:E6.1 433 TMEM14C chr6:10728727-10756467(+)
GIHPSKVVY,ENSG00000263809:E3.1-E4.1 432 unknown_gene chr17:8376104-8379796(-)
RPYLPVKVL,ENSG00000134330:E8.4-E9.1 432 IAH1 chr2:9484550-9487456(+)
LPPPRLASV,ENSG00000090581:E5.10-E5.24 428 GNPTG chr16:1362320-1362452(+)
SSQVHLSHL,ENSG00000172053:E11.8-E11.11 425 QARS1 chr3:49099853-49099994(-)